专业背景:计算机科学 研究方向与兴趣: JavaEE-Web软件开发, 生物信息学, 数据挖掘与机器学习, 智能信息系统 目前工作: 基因组, 转录组, NGS高通量数据分析, 生物数据挖掘, 植物系统发育和比较进化基因组学
http://liucheng.name/770/
序列的Fasta格式是最经常看到的格式之一。下面简介说明一下什么是FASTA格式。
Fasta格式开始于一个标识符:">",然后是一行描述,下面是一行行的序列。每一行最好不要超过80个字母。
如:
>gi|532319|pir|TVFV2E|TVFV2E envelope protein
ELRLRYCAPAGFALLKCNDADYDGFKTNCSNVSVVHCTNLMNTTVTTGLLLNGSYSENRT
QIWQKHRTSNDSALILLNKHYNLTVTCKRPGNKTVLPVTIMAGLVFHSQKYNLRLRQAWC
HFPSNWKGAWKEVKEEIVNLPKERYRGTNDPKRIFFQRQWGDPETANLWFNCHGEFFYCK
MDWFLNYLNNLTVDADHNECKNTSGTKSGNKRAPGPCVQRTYVACHIRSVIIWLETISKK
TYAPPREGHLECTSTVTGMTVELNYIPKNRTNVTLSPQIESIWAAELDRYKLVEITPIGF
APTEVRRYTGGHERQKRVPFVXXXXXXXXXXXXXXXXXXXXXXVQSQHLLAGILQQQKNL
LAAVEAQQQMLKLTIWGVK
下面再说一下每个字母或字符所代表的含义。
A --> adenosine M --> A C (amino)
C --> cytidine S --> G C (strong)
G --> guanine W --> A T (weak)
T --> thymidine B --> G T C
U --> uridine D --> G A T
R --> G A (purine) H --> A C T
Y --> T C (pyrimidine) V --> G C A
K --> G T (keto) N --> A G C T (any)
- gap of indeterminate length
A alanine P proline
B aspartate or asparagine Q glutamine
C cystine R arginine
D aspartate S serine
E glutamate T threonine
F phenylalanine U selenocysteine
G glycine V valine
H histidine W tryptophan
I isoleucine Y tyrosine
K lysine Z glutamate or glutamine
L leucine X any
M methionine * translation stop
N asparagine - gap of indeterminate length
转载注明 : 来源于 柳城博客
Posted on 22 七月 2009 by Lc. ,阅读 197
看清楚,这里所说的是Fastq格式,不是Fasta格式,要了解Fasta格式,请看Fasta格式的详细说明。Fastq格式也是序列格式中常见的一种。下面简单介绍一下FASTQ格式,
A FASTQ file normally uses four lines per sequence. Line 1 begins with a '@' character and is followed by a sequence identifier and an optional description (like a FASTA title line). Line 2 is the raw sequence letters. Line 3 begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again. Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
FASTQ格式的序列一般都包含有四行,第一行由'@'开始,后面跟着序列的描述信息,这点跟FASTA格式是一样的。第二行是序列。第三行由'+'开始,后面也可以跟着序列的描述信息。第四行是第二行序列的质量评价(quality values,注:应该是测序的质量评价),字符数跟第二行的序列是相等的。
FASTQ格式例子:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
例如在NCBI看到的FASTQ格式如下:
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
至于序列的quality values值,是通过一些算法得出来的。具体也搞不明白,不多讲。另外FASTQ格式是不至一种的,不同的来源会有些差异,如Illumina 1.0 FASTQ 、 Sanger FASTQ等。都是比较特殊的情况。
FASTQ格式与Fasta格式、GenBank等格式的相互转换,看BioPerl指南 – 序列格式的转换
转自柳城博客:http://liucheng.name/825/
FASTQ format is a text-based format for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are encoded with a single ASCII character for brevity. It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA sequence and its quality data, but has recently become the de facto standard for storing the output of high throughput sequencing instruments such as the Illumina Genome Analyzer.[1]
A FASTQ file normally uses four lines per sequence.
A FASTQ file containing a single sequence might look like this:
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
The character '!' represents the lowest quality while '~' is the highest. Here are the quality value characters in left-to-right increasing order of quality (ASCII):
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
The original Sanger FASTQ files also allowed the sequence and quality strings to be wrapped (split over multiple lines), but this is generally discouraged as it can make parsing complicated due to the unfortunate choice of "@" and "+" as markers (these characters can also occur in the quality string).
Sequences from the Illumina software use a systematic identifier:
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | the unique instrument name |
|---|---|
| 6 | flowcell lane |
| 73 | tile number within the flowcell lane |
| 941 | 'x'-coordinate of the cluster within the tile |
| 1973 | 'y'-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
Versions of the Illumina pipeline since 1.4 appear to use #NNNNNN instead of #0 for the multiplex ID, where NNNNNN is the sequence of the multiplex tag.
With Casava 1.8 the format of the '@' line has changed:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | the unique instrument name |
|---|---|
| 136 | the run id |
| FC706VJ | the flowcell id |
| 2 | flowcell lane |
| 2104 | tile number within the flowcell lane |
| 15343 | 'x'-coordinate of the cluster within the tile |
| 197393 | 'y'-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y | Y if the read fails filter (read is bad), N otherwise |
| 18 | 0 when none of the control bits are on, otherwise it is an even number |
| ATCACG | index sequence |
FASTQ files from the NCBI/EBI Sequence Read Archive often include a description, e.g.
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
In this example there is an NCBI-assigned identifier, and the description holds the original identifier from Solexa/Illumina (as described above) plus the read length.
Also note that the NCBI have converted this FASTQ data from the original Solexa/Illumina encoding to the Sanger standard (see encodings below).
A quality value Q is an integer mapping of p (i.e., the probability that the corresponding base call is incorrect). Two different equations have been in use. The first is the standard Sanger variant to assess reliability of a base call, otherwise known as Phred quality score:
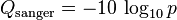
The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds p/(1-p) instead of the probability p:

Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q < 13).

At times there has been disagreement about which mapping Illumina
actually uses. The user guide (Appendix B, page 122) for version 1.4 of
the Illumina pipeline states that: "The scores are defined as
Q=10*log10(p/(1-p)) [sic], where p is the probability of a base call corresponding to the base in question".[2]
In retrospect, this entry in the manual appears to have been an error.
The user guide (What's New, page 5) for version 1.5 of the Illumina
pipeline lists this description instead: "Important Changes in Pipeline
v1.3 [sic].
The quality scoring scheme has changed to the Phred [i.e., Sanger]
scoring scheme, encoded as an ASCII character by adding 64 to the Phred
value. A Phred score of a base is:  , where e is the estimated probability of a base being wrong.[3]
, where e is the estimated probability of a base being wrong.[3]
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT +HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
An alternative interpretation of this ASCII encoding has been proposed.[8] Also, in Illumina runs using PhiX controls, the character 'B' was observed to represent an "unknown quality score". The error rate of 'B' reads was roughly 3 phred scores lower the mean observed score of a given run.
For raw reads, the range of scores will depend on the technology and the base caller used, but will typically be up to 41 for recent Illumina chemistry. Since the maximum observed quality score was previously only 40, various scripts and tools break when they encounter data with quality values larger than 40. For processed reads, scores may be even higher. For example, quality values of 45 are observed in reads from Illumina's Long Read Sequencing Service (previously Moleculo).
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX...................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... .................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ...................... ..LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ | | | | | | 33 59 64 73 104 126 0........................26...31.......40 -5....0........9.............................40 0........9.............................40 3.....9.............................40 0.2......................26...31........41 S - Sanger Phred+33, raw reads typically (0, 40) X - Solexa Solexa+64, raw reads typically (-5, 40) I - Illumina 1.3+ Phred+64, raw reads typically (0, 40) J - Illumina 1.5+ Phred+64, raw reads typically (3, 40) with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold) (Note: See discussion above). L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
For SOLiD data, the sequence is in color space, except the first position. The quality values are those of the Sanger format. Alignment tools differ in their preferred version of the quality values: some include a quality score (set to 0, i.e. '!') for the leading nucleotide, others do not. The sequence read archive includes this quality score.
Quality values account for about half of the required disk space in the FASTQ format (before compression), and therefore the compression of the quality values can significantly reduce storage requirements and speed up analysis and transmission of sequencing data. Both lossless and lossy compression are recently being considered in the literature. For example, the algorithm QualComp [9] performs lossy compression with a rate (number of bits per quality value) specified by the user. Based on rate-distortion theory results, it allocates the number of bits so as to minimize the MSE (mean squared error) between the original (uncompressed) and the reconstructed (after compression) quality values. Other algorithms for compression of quality values include SCALCE [10] and Fastqz.[11] Both are lossless compression algorithms that provide an optional controlled lossy transformation approach. For example, SCALCE reduces the alphabet size based on the observation that “neighboring” quality values are similar in general.
There is no standard file extension for a FASTQ file, but .fq and .fastq, are commonly used.
FASTQ to FASTA format:
zcat input_file.fastq.gz | awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' > output_file.fa
Illumina FASTQ 1.8 to 1.3
sed -e '4~4y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJ/@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\\]^_`abcdefghi/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.3 to 1.8
sed -e '4~4y/@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\\]^_`abcdefghi/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJ/' myfile.fastq # add -i to save the result to the same input file

评论